All examples in this tutorial were generated by Flux Simulator with this script from the Rail repo.
Trial and error
We made a serious attempt to design Rail-RNA for people who don't read manuals to discourage improper use of the software. In general, you can get started by entering
rail-rna
in the shell. You'll obtain the following output, perhaps with a different version number.
error: too few arguments
usage: rail-rna <job flow> <mode> <[args]>
<job flow> {prep, align, go}
prep: preprocess reads listed in a required manifest
file (specified with --manifest)
align: align preprocessed reads (specified with --input)
go: perform prep and align in succession
<mode> {local, parallel, elastic}
local: run Rail-RNA on this computer
parallel: run Rail-RNA on all active IPython engines
elastic: run Rail-RNA on Amazon Elastic MapReduce.
Requires that the user sign up for Amazon Web Services
∀ Rail-RNA v0.2.0 by Abhi Nellore (anellore@jhu.edu; nellore.github.io)
Rail-RNA is a scalable MapReduce pipeline that can analyze many RNA-seq
datasets at once. To view help for a given combination of <job flow> and
<mode>, specify both, then add -h/--help.
It's quite possible to figure out how a substantial fraction Rail-RNA works by trial and error. Follow the prompts and fix your errors, which Rail tries to explain in detail when they're encountered. Please let us know in the Gitter if you're confused by any errors output. If you prefer to be guided through some examples, read on.
Modes and job flows
Rail-RNA has three modes---
-
local, which runs the software on a single machine on up to as many processing cores as available. By default, Rail-RNA runs on the number of processing cores it detects on a machine less one to avoid monopolizing a single machine's resources. To adjust the number of cores on which Rail is run, use the-p/--num-processescommand-line parameter described under Reference. -
elastic, which runs the software on an Amazon Elastic MapReduce computer cluster in the cloud. This costs money: you rent computing capacity (on Elastic Compute Cloud, or EC2) and storage (on Simple Storage Service, or S3) from Amazon for the duration of your job flow and for as long as you keep your outputs in the cloud. Price information is here. For a medium-size job (up to ~100 RNA-seq samples with ~50 million reads each), we tend to use a cluster of 40 c3.2xlarge machines, which costs $21/hour on demand but perhaps less than half that number on the spot market in the US Standard zone (us-east-1). Learn more about the spot market and bidding on compute capacity here. -
parallel, which runs on an IPython Parallel cluster whose profile is specified at the command line. You must set up the IPython Parallel cluster yourself.
---and three job flows---
-
prep, which if necessary downloads input FASTQ/FASTA files from a remote server and casts it in a form amenable for further analysis. This job flow must always be run before thealignjob flow. -
align, which aligns preprocessed data and writes outputs explained in Deliverables. Use the--deliverablesoption described there to control which of Rail-RNA's terminal outputs should be written. -
go, which runs theprepandalignjob flows in succession.
To run some combination of mode and job flow, start your command with
rail-rna <mode> <job flow>
. Get help by entering
rail-rna <mode> <job flow> -h
The reason for separating the prep and align job flows is that you may prefer to run these flows on different numbers of processing cores: if downloads are throttled by the remote server, and only a small number of threads can download input files concurrently, then you can limit the size of the computer cluster in elastic mode (with the -c/--core-instance-count parameter described in Reference) or the number of concurrently running threads in local mode (with the -p/--num-processes parameter described in Reference).
Align some RNA-seq samples with us in local, parallel, and elastic modes below. We assume your login name is testuser, and your home directory is /home/testuser.
local and parallel modes: Drosophila examples
Rail-RNA wraps both Bowtie 1 and Bowtie 2, so you will need both Bowtie 1 and Bowtie 2 indexes of a genome assembly to perform alignment. It's easiest to download one of the Illumina iGenomes, collections of reference sequences and indexes for popularly studied organisms. Here are explicit instructions:
Download the Drosophila melanogaster build dm3 iGenome, available here. This is a 783-MB file. We assume you downloaded the dm3 iGenome to the directory /home/testuser/Downloads. Unpack it by entering
cd /home/testuser/Downloads
tar xvzf Drosophila_melanogaster_UCSC_dm3.tar.gz
in the shell. This could take a while, but once it's done, the Bowtie 1 index basename should be /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/BowtieIndex/genome, while the Bowtie 2 index basename should be /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/Bowtie2Index/genome.
Warning: You may think you already have Bowtie 1 and 2 indexes for a given genome assembly---perhaps dm3---but the contigs in one may not be exactly the same as the contigs in the other. When working with any reference in Rail-RNA, it is important to ensure that the Bowtie 1 and 2 indexes are built from the same FASTA. Check the Bowtie 1 documentation and Bowtie 2 documentation for information on how to build Bowtie indexes from reference FASTAs.
Now open your browser, and navigate to this URL. You'll see the following text.
http://verve.webfactional.com/dm3_example_1_left.fastq 0 http://verve.webfactional.com/dm3_example_1_right.fastq 0 dm3_example-1-1
http://verve.webfactional.com/dm3_example_2_left.fastq 0 http://verve.webfactional.com/dm3_example_2_right.fastq 0 dm3_example-2-1
This is a Rail-RNA manifest file, and it is required for all Rail-RNA runs. Its format mirrors the manifest file format of Myrna, a predecessor of Rail. Each line corresponds to a different RNA-seq sample. A line for a single-end sample looks like this---
<FASTQ/FASTA URL>(tab)<URL MD5 checksum or 0>(tab)<sample label>
---while a line for a paired-end sample looks like this---
<FASTQ/FASTA URL 1>(tab)<URL 1 MD5 checksum or 0>(tab)<FASTQ/FASTA URL 2>(tab)<URL 2 MD5 checksum or 0>(tab)<sample label>
. URLs can be on the local filesystem, on the web, or if the AWS CLI is installed, on Amazon S3. If an input file is not on the local filesystem, it will be downloaded by Rail. Manifest files may be hosted remotely as well.
Our Drosophila example is composed of two paired-end biological replicates, and the raw data is hosted at http://verve.webfactional.com . Let's create a new railtests directory in testuser's home directory for running all our examples.
mkdir -p /home/testuser/railtests
cd /home/testuser/railtests
Preprocessing and aligning the test data in local mode takes a single command:
rail-rna go local -x /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/BowtieIndex/genome /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/Bowtie2Index/genome -m https://raw.githubusercontent.com/nellore/rail/master/ex/dm3_example.manifest
. The run should complete after a few minutes. You can browse the output by entering
cd /home/testuser/railtests/rail-rna_out
ls
You'll find four directories:
alignments cross_sample_results
coverage_bigwigs junctions_and_indels
See Deliverables for information on how to interpret what's in these directories.
Commonly used options in local mode are
-p/--num-processes, which controls the number of processes Rail-RNA runs simultaneously. By default, Rail uses as many processing cores as your computer has less one so it doesn't monopolize resources. You may want to run on all cylinders by setting this equal to the number of available processing cores.
-o/--output, which changes the output directory. By default, this is rail-rna_out in the current directory.
See the Reference section for a comprehensive description of command-line parameters.
We could have run Rail-RNA in two steps: by preprocessing first and subsequently aligning. You can try this yourself by running the following commands in sequence.
cd /home/testuser/railtests
rail-rna prep local -m https://raw.githubusercontent.com/nellore/rail/master/ex/dm3_example.manifest -o dmel_prepped -p 1
rail-rna align local -i dmel_prepped -m https://raw.githubusercontent.com/nellore/rail/master/ex/dm3_example.manifest -x home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/BowtieIndex/genome /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/Bowtie2Index/genome -f
The rail-rna prep local command above is executed on a single thread (with -p 1) to ensure that only one download is performed at a time, which would be useful if our downloads were throttled. In the rail-rna align local command, the parameter -f/--force tells Rail to overwrite any existing outputs. You should end up with exactly the same output you obtained when you ran the rail-rna go local command.
Now you'll run an example that will break Rail. Navigate to this URL, a manifest file that should look like this:
http://verve.webfactional.com/bad.fastq 0 bad-1-1
It's a single-end sample called "bad," presumably because it's not good. Visit http://verve.webfactional.com/bad.fastq next. The file will be downloaded, or it will be displayed in your browser. If it's downloaded, navigate to the appropriate folder at the shell prompt, and enter
cat bad.fastq
. Either way, you'll see
@badrecord
ATACAGATGACAGATGACAGGGTAGAGACAAATAGACAGATGACGATGGACAGATGACAGATAGAACAGATAGAGA
+
IIIIIIIIIIIIII
@goodrecord
ATGGCATCAGTCAAGTCAAGATTACTAGTAGCCATACAAGATACATCGTTTAACGATTGTGGCACATACGTCACCA
+
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII
There's one sequence labeled "badrecord" and another labeled "goodrecord". "badrecord" is bad because its read sequence isn't the same length as its quality sequence. Without being told otherwise, Rail-RNA chokes on bad records, but it's good to see how it chokes to learn how to diagnose problems. Run Rail-RNA on the bad manifest file like so---
cd /home/testuser/railtests
rail-rna go local -x /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/BowtieIndex/genome /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/Bowtie2Index/genome -m https://raw.githubusercontent.com/nellore/rail/master/ex/bad.manifest -o ./bad
---and you'll ultimately obtain output like this:
∀ Rail-RNA v0.2.0
Started job flow on Saturday, Jul 18, 2015 at 07:36:29 PM EDT.
~.oOo.>
00h:00m:00s |___| Step 1/24: Count lines in input files
00h:00m:01s |___| Completed 1 task.
00h:00m:01s |___| Deleted temporary files.
00h:00m:01s |___| Step 2/24: Assign reads to preprocessing tasks
00h:00m:01s |___| Partitioned 1 input into tasks.
00h:00m:03s |___| Completed 1 task.
00h:00m:03s |___| Deleted temporary files.
00h:00m:03s |___| Step 3/24: Preprocess reads
*****Errors encountered*****
Streaming command "cat /var/folders/rs/zytvc1753p9_bth4lrhbm7c80000gn/T/tmpCxbBKB/0 | /usr/local/raildotbio/pypy-2.5.0-osx64/bin/pypy /usr/local/raildotbio/rail-rna/rna/steps/preprocess.py --nucs-per-file=100000000 --gzip-output --push=/home/testuser/railtests/rail-rna_logs/preprocess/push --gzip-level 3 --bin-qualities >/home/testuser/railtests/rail-rna_logs/preprocess/0 2>/home/testuser/railtests/rail-rna_logs/preprocess/dp.map.log/0.0.log" failed; exit level was 1.
Job flow failed on Saturday, Jul 18, 2015 at 07:36:34 PM EDT. Run time was 5.503 seconds.
To start this job flow from where it left off, run:
/usr/local/raildotbio/pypy-2.5.0-osx64/bin/pypy /usr/local/raildotbio/rail-rna/dooplicity/emr_simulator.py -j /home/testuser/railtests/rail-rna_logs/resume_flow_WDEZ24GVC0TM.json -b /usr/local/raildotbio/rail-rna/rna/driver/rail-rna.txt -l /home/testuser/railtests/rail-rna_logs/flow.2015-07-18T19:36:27.934937.log -f --max-attempts 1 --num-processes 3
Traceback (most recent call last):
File "app_main.py", line 75, in run_toplevel
File "/usr/local/raildotbio/rail-rna/dooplicity/emr_simulator.py", line 1963, in <module>
args.scratch, args.common, args.sort, args.max_attempts)
File "/usr/local/raildotbio/rail-rna/dooplicity/emr_simulator.py", line 1719, in run_simulation
max_attempts=max_attempts
File "/usr/local/raildotbio/rail-rna/dooplicity/emr_simulator.py", line 1290, in execute_balanced_job_with_retries
raise RuntimeError
RuntimeError
Rail is telling us that something went wrong with a command during preprocessing. Note the following.
-
Rail gives you a command you can use to resume a job flow if it's failed for a fixable reason. One possible reason is that you ran out of space on disk during a job flow, and you need to delete some of your files. You can highlight and copy the command that resumes your job flow with
Command+Con a Mac orCTRL+Cin Linux. If somehow you lose the resume command, you'll also find it in the last written file whose extension is.login the log directory/home/testuser/railtests/rail-rna_logs. Enterls -tr /home/testuser/railtests/rail-rna_logs/*.log | tail -n 1to get its path.
-
There's a log file in the command that failed, right after the
2>. This file will tell you why the job flow failed. Open it withless:less /home/testuser/dmel/rail-rna_logs/preprocess/dp.map.log/0.0.log. (You should
lesswhatever log file appears for you after the2>). At the bottom of the file is the exception that made the job flow fail:Created local destination directory "/var/folders/rs/zytvc1753p9_bth4lrhbm7c80000gn/T/tmpQJHduB". Retrieving URL "http://verve.webfactional.com/bad.fastq"... Range of quality values found from random sample of 10000 records is (73, 73). Guessed Phred64 encoding. Traceback (most recent call last): File "app_main.py", line 75, in run_toplevel File "/usr/local/raildotbio/rail-rna/rna/steps/preprocess.py", line 863, in <module> mover=mover) File "/usr/local/raildotbio/rail-rna/rna/steps/preprocess.py", line 557, in go ) % (line_numbers[i], sources[i]) AssertionError: Length of read sequence does not match length of quality string at line 4 of file "/var/folders/rs/zytvc1753p9_bth4lrhbm7c80000gn/T/tmpQJHduB/bad.fastq".. So Rail noticed
@badrecordwas a bad record and failed. You can have Rail ignore bad records during preprocessing using the--ignore-bad-recordscommand-line parameter:rail-rna go local -x /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/BowtieIndex/genome /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/Bowtie2Index/genome -m https://raw.githubusercontent.com/nellore/rail/master/ex/bad.manifest -o ./bad -f --ignore-bad-records. This job flow should succeed, and you'll end up with exactly one aligned read in the directory
/home/testuser/railtests/bad/alignments. Ignoring bad records may be useful when you're analyzing hundreds of samples and, for example, one file is truncated, but you'd like to analyze all its available reads anyway. Sometimes, you can't anticipate the integrity of the data you're analyzing, but you don't want your job flow to fail because of a handful of bad records.
To test Rail-RNA in parallel mode, you should have IPython installed. If the Rail-RNA installer didn't prompt you to install it, you already have it. Detailed instructions on starting IPython clusters over many different cluster configurations may be found here. The IPython documentation around this link teaches you how to create a profile for a cluster configuration and how to start a cluster. An IPython cluster is nothing but a collection of Python interpreters ("engines") running on processing cores distributed across some networked computers. Rail-RNA does not start an IPython cluster for you; rather, it detects a running IPython cluster and runs itself over that. You can choose the IPython profile Rail-RNA should use with the --ipython-profile command-line parameter. If left unspecified, this is taken to be the default profile.
For testing purposes, you can run an IPython cluster on just your machine by entering
ipcluster start -n <k>
, where <k> is the number of IPython engine processes you want to run. <k> is analogous to the -p/--num-processes parameter of Rail-RNA in local mode. Now rerun the first Drosophila example except in parallel mode by entering
rail-rna go parallel -x /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/BowtieIndex/genome /home/testuser/Downloads/Drosophila_melanogaster/UCSC/dm3/Sequence/Bowtie2Index/genome -m https://raw.githubusercontent.com/nellore/rail/master/ex/dm3_example.manifest -f
What happens is pretty unimpressive: Rail runs like it does in local mode. But IPython Parallel lends Rail versatility: there are many different IPython cluster configurations you can set up on, say, your institutional computer cluster. But there is one caveat: in parallel mode, you must make sure the output directory (specified with -o/--output) and the log directory (specified with --log) are at paths accessible to all the nodes in your cluster running IPython engines. Rail-RNA aggregates intermediate data in the log directory between any two successive steps, and all nodes must be able to stream data from that directory to complete their assigned tasks. Moreover, outputs must go to the same place, and some paths on a cluster refer to different places on different nodes. For example, you probably shouldn't make a subdirectory of /tmp your output or log directory on a conventional computer cluster because /tmp tends to be a node-local temporary directory.
elastic mode: a human example
Warning: running this example will cost money, but probably no more than US$5. Read our disclaimer before using elastic mode.
You should have performed these steps to set up the AWS CLI and Elastic MapReduce before attempting the example.
Check out this manifest file in your browser. Listed are two single-end human samples with just 20,000 reads each. They were generated with expression profiles of two GEUVADIS lymphoblastoid cell line samples in a way we describe in our paper, but because there are so few reads, you probably couldn't tell which GEUVADIS samples we used if you didn't have the sample labels in the manifest file.
Let's use Elastic MapReduce to both preprocess and align these data. Since you'll be storing the results on Amazon's Simple Storage Service (S3), you should first read up on working with buckets. The short story is that on S3, a bucket is something like a directory in a filesystem, except
- Unless you specify otherwise, only your account can access it.
- Bucket names are globally unique: if someone else has taken a bucket name in any Amazon region, you can't have it.
- Underscores in bucket names are a bad idea when you use Elastic MapReduce. They cause weird problems. Don't ask why; just go with it.
- A bucket is located in a specific Amazon region---that is, a bucket has a physical location in an Amazon data center, and that's where your files go.
You can create a bucket at the command line using the AWS CLI. Enter
aws s3 mb s3://this-is-the-bucket-name-you-make-up --region <a valid region>
. A list of valid regions is available here. Since you specified your default region when configuring the AWS CLI, if you leave the --region part out above, the bucket will be created in your default region---typically us-east-1.
But you don't have to create a bucket yourself before using Rail; if it doesn't already exist, Rail will automatically create the bucket you specify in your output directory path for you. We went through this exercise so you understand that the root of your output directory on S3 will always be a bucket in some region. Note that you can use Amazon's web interface to manipulate buckets, too.
If you've set up the AWS CLI properly, you can preprocess and align the example human dataset in the cloud with one command:
rail-rna go elastic -m https://raw.githubusercontent.com/nellore/rail/master/ex/hg19_example.manifest -a hg19 -o s3://this-is-the-bucket-name-you-make-up/human_example --core-instance-type c3.2xlarge --master-instance-type c3.2xlarge -c 1
. This command reserves two c3.2xlarge EC2 instances to execute a Rail-RNA job flow that dumps its output to s3://this-is-the-bucket-name-you-make-up/human_example. =There's always one master instance, whose type is specified with the --master-instance-type command-line parameter above; and the -c parameter specifies the number of core instances, whose type is specified with --core-instance-type. The master instance manages the Hadoop cluster that will run your job, scheduling and coordinating tasks that are executed on slave nodes---which for Rail-RNA are generally core instances. We recommend using at least 40 c3.2xlarge core instances for every hundred RNA-seq samples with 50 million reads each. You might also consider using c3.8xlarge instances, each of which has four times the resources of a c3.2xlarge instance; so in this case, the recommended ratio is at least 10 c3.8xlarge instances for every hundred RNA-seq samples with 50 million reads each. For our small example, we use only one core instance. The -a hg19 parameter specifies that the input data should be aligned to hg19.
Warning: Your job flow will execute in the default region you chose when you set up the AWS CLI unless you tack a --region <desired region> onto the command above. If your output bucket is in a region different from your Elastic MapReduce cluster, transferring data between the cluster and S3 will take longer and slow down your job.
Executing the rail-rna go elastic ... command above gives the following output.
testcomputer:~ testuser$ rail-rna go elastic -m https://raw.githubusercontent.com/nellore/rail/master/ex/hg19_example.manifest -a hg19 -o s3://this-is-the-bucket-name-you-make-up/human_example --core-instance-type c3.2xlarge --master-instance-type c3.2xlarge -c 1
Loading...
Checked all files listed in manifest file.
Copied Rail-RNA and bootstraps to S3.
∀ Rail-RNA v0.2.0
Started job flow submission script on Saturday, Jul 18, 2015 at 08:50:34 PM EDT.
~.oOo.>
00h:00m:00s |___| Read job flow from input JSON.
00h:00m:10s |___| Verified that output directories on S3 are writable.
00h:00m:23s |___| Set up output directories on S3.
00h:00m:25s |___| Submitted job flow.
*****Job flow ID is j-2NSJ3SJPGKC58 .*****
*****Submission can be monitored at https://console.aws.amazon.com/elasticmapreduce/?region=eu-west-1#cluster-details:j-2NSJ3SJPGKC58 .*****
00h:00m:25s |___| Opening URL in default browser, if possible.
<.oOo.~
Finished job flow submission script on Saturday, Jul 18, 2015 at 08:51:00 PM EDT. Run time was 25.789 seconds.
If all goes well, your browser will open the Elastic MapReduce interface for monitoring your job flow. If that doesn't happen, the URL for viewing your job flow is included in Rail-RNA's output, and you can copy and paste it into your browser's address bar.
Explore. Click on things. You'll first see something like
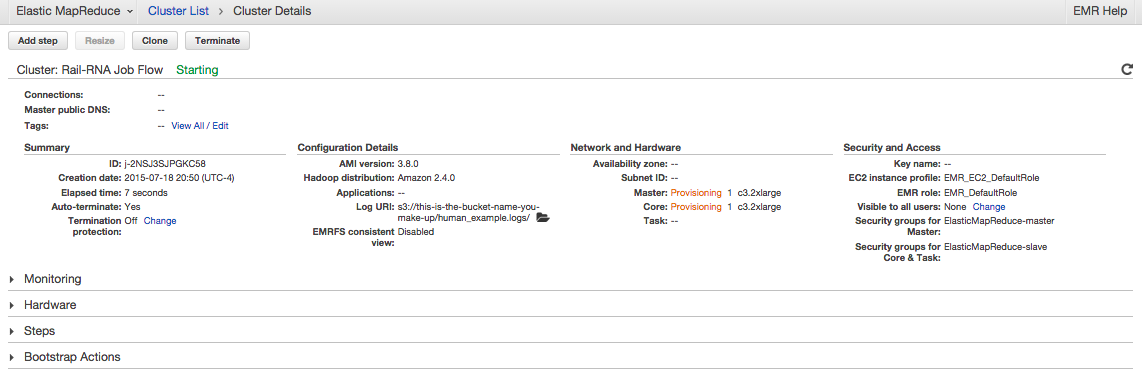
. If you click on Steps, you'll see
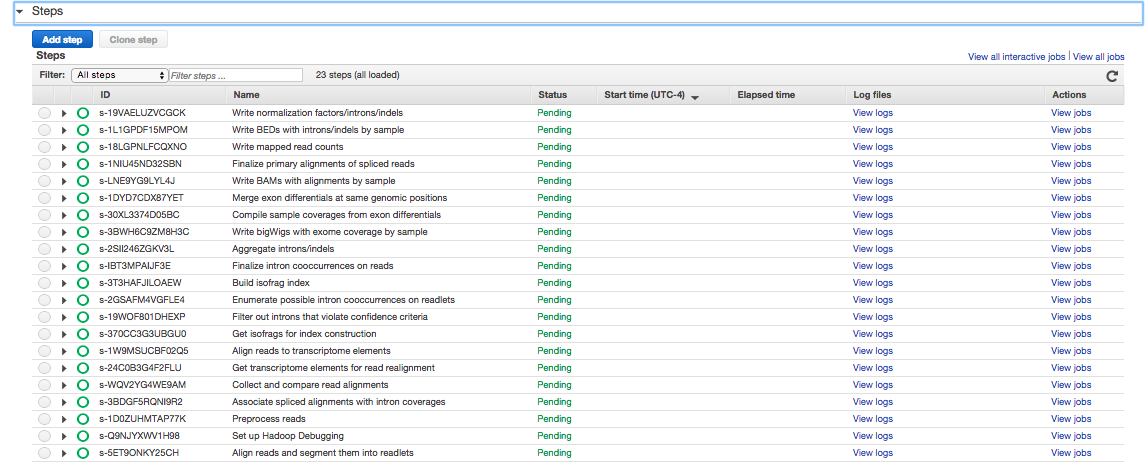
. These are the steps Rail-RNA will march through to align the human example. If you click on Bootstrap Actions, you'll see
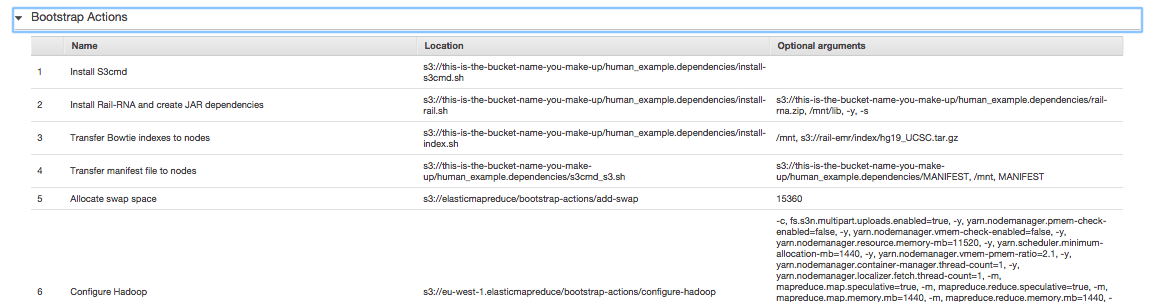
. When an Elastic MapReduce cluster starts up, its nodes are not equipped with the software Rail needs to run a job flow. Bootstrap actions are executed before the job flow begins to install this required software. Once the job flow is finished, all the software and any temporary files that have accumulated on the cluster are wiped. This is a virtue of using cloud computing to do your bioinformatics: you start with exactly the same machines and software configuration, making your results highly reproducible.
It's possible you'll want to click on Terminate soon after starting the job flow to avoid incurring any charges---and that's fine, but you may also want to delete Rail-RNA's detritus on S3 using the console. This includes the directories s3://this-is-the-bucket-name-you-make-up/human_example.dependencies and s3://this-is-the-bucket-name-you-make-up/human_example.logs. When Rail-RNA launches a job on Elastic MapReduce, it copies itself to S3 so the version of Rail you use in the cloud is precisely the version you use on your computer. This copy is stored in the directory that ends with dependencies. The directory that ends with logs is used by Elastic MapReduce to record stats on your job flow. This is what you view in the Elastic MapReduce web interface. Another directory---s3://this-is-the-bucket-name-you-make-up/human_example.intermediate---will appear on S3 if you run the job flow from end to end. This directory stores intermediate data from one step that's streamed into some other step of the Rail-RNA pipeline. It shouldn't be touched until a job flow is complete, but afterwards, feel free to axe it. Both the intermediate and dependencies directories are purged automatically after four days to avoid your incurring extra S3 charges without your noticing. You can toggle how many days these directories remain on S3 with the --intermediate-lifetime command-line parameter.
Here's a screenshot of the job flow in progress.
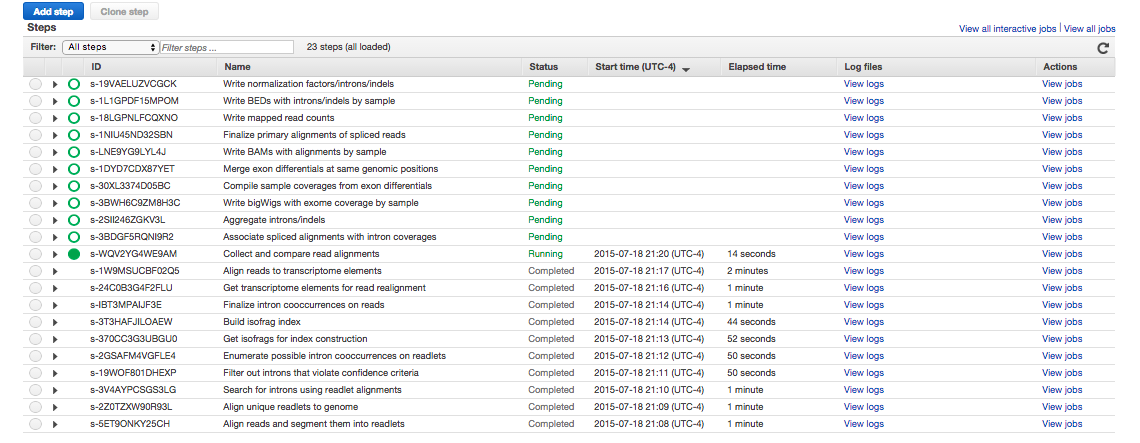
If you decided to continue the job flow, and your steps don't look something like this, something's gone wrong. Can't figure out what? Complain in the Gitter.
When the job flow is complete---
---you can browse the outputs in the console. You'll find them at s3://this-is-the-bucket-name-you-make-up/human_example. The AWS CLI can also be used to list the contents of a directory like so:
testcomputer:~ testuser$ aws s3 ls s3://this-is-the-bucket-name-you-make-up/human_example/
PRE alignments/
PRE coverage_bigwigs/
PRE cross_sample_results/
PRE junctions_and_indels/
You can download all the results to your computer like so:
mkdir /home/testuser/human_example
cd /home/testuser/human_example
aws s3 cp s3://this-is-the-bucket-name-you-make-up/human_example/ ./ --recursive
. Warning: transfers from S3 to non-EC2 computers cost money.
To divide the human example up into preprocess and align job flows, try the following commands.
rail-rna prep elastic -m https://raw.githubusercontent.com/nellore/rail/master/ex/hg19_example.manifest -o s3://this-is-the-bucket-name-you-make-up/human_example --core-instance-type c3.2xlarge --master-instance-type c3.2xlarge -c 1
rail-rna align elastic -m https://raw.githubusercontent.com/nellore/rail/master/ex/hg19_example.manifest -a hg19 -o s3://this-is-the-bucket-name-you-make-up/human_example --core-instance-type c3.2xlarge --master-instance-type c3.2xlarge -c 1
You should now know enough figure out how to use Rail-RNA to align your RNA-seq data! Refer to the Deliverables and Reference for further details on, respectively, changing output formats and command-line parameters. To set up Elastic MapReduce for analyzing dbGaP-protected RNA-seq data with Rail-RNA, refer to dbGaP on EMR. Note that the instructions there may require that you contact your AWS administrator.